Гиперкератоз (hyperkeratosis)
— чрезмерное утолщение рогового слоя эпидермиса.
Гиперкератоз может быть результатом избыточного образования кератина, когда зернистый и шиповатый слои утолщаются (пролиферационный гиперкератоз), как, например, при красном плоском лишае, а также следствием задержки отторжения роговых клеток (ретенционный гиперкератоз), при котором зернистый, а иногда и шиповатый слои истончаются, например при ихтиозе.
В основе пролиферационного гиперкератоза лежит интенсивный синтез кератина при повышенной функциональной активности клеток эпидермиса, а в основе ретенционного — повышение содержания в роговом слое мукополисахаридов, играющих цементирующую роль, мешающих разъединению клеток рогового слоя при шелушении.
«Патоморфологическая диагностика заболеваний кожи»,
Г.М.Цветкова, В.К.Мордовцев
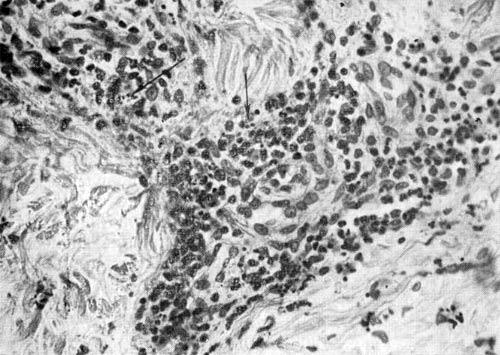
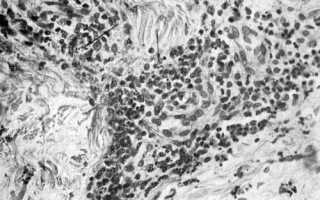
Лейкоклазия (кариорексис) — дезинтеграция ядер зернистых лейкоцитов, приводящая к образованию ядерной «пыли» (темного цвета), патогномонична для аллергического васкулита. Лейкоклазия Лейкоклазия (показано стрелками) зернистых лейкоцитов при аллергическом васкулите. X 250. Метахромазия — феномен изменения окраски. Метахромазия соединительной ткани может быть вызвана наличием в ней гликозаминогликанов или амилоида. Гликозаминогликаны окрашиваются метиленовым или толуидиновым сипим в пурпурный цвет,…
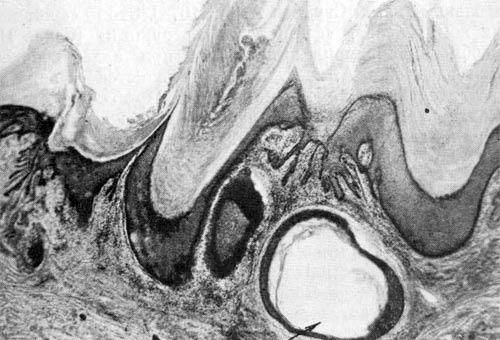
Слизистая дистрофия может появляться при патологии как в эпидермисе, так и собственно коже. Дермальный муцин в норме формирует основную субстанцию соединительной ткани и состоит из гликозаминогликанов, преимущественно гиалуроновой кислоты. Это вид муцина — ШИК-отрицательный, окрашивается альциановым синим при рН 2,5 — 0,4, метахроматично окрашивается метиленовым синим и толуидиновым синим, гиалуронидазолабилен. Эпителиальный муцин, называемый сиаломуцином, содержит…
Фибриноидное изменение соединительной ткани дермы представляет собой результат глубокой дезорганизации ее, в основе которого лежит деструкция коллагена и межуточного вещества с образованием белково-полисахаридных комплексов, не встречающихся в нормальных условиях, и появлением фибриноида. Фибриноидно измененные участки резко эозинофильны, часто гомогенны. В начале процесса в этих участках выявляются гликозаминогликаны, метахроматично окрашивающиеся толуидиновым синим, позднее — нейтральные МПС….
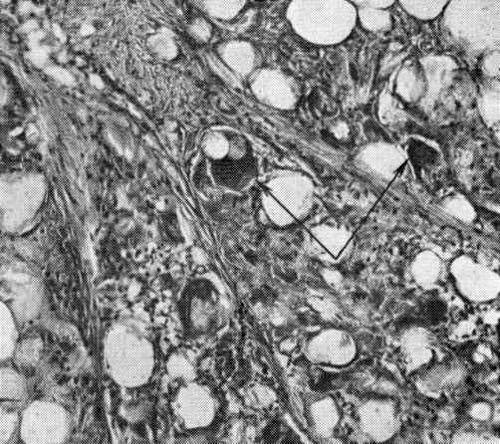

Грануляционная ткань образуется в ранах и язвах, а также при хронических воспалительных процессах в коже. Состоит из новообразованных капилляров и клеток, среди которых большое число фибробластов. Между капиллярами, как правило, располагается воспалительный инфильтрат, состоящий из лимфоидных клеток, макрофагоцитов и плазмоцитов. Волокнистые структуры появляются позже. Гранулематозные структуры Гранулематозные структуры в подкожной клетчатке с наличием в них…
У животных, как и у человека, встречается множество кожных заболеваний определяемых наследственными факторами, наследственной предрасположенностью. К таким болезням относятся генодерматозы, склеродермия, атопический дерматит и др.
Генодерматозы.
Генодерматозы – наследственные заболевания кожи, объединяющие множество нозологических форм, проявляющихся различными патологическими процессами. У человека наиболее часто встречается ихтиоз (вульгарный, Х-сцепленный, ихтиозиформная эритродермия), кератодермия, буллёзный эпидермолиз (О. Л.Иванов,2002).У собак и других животных из группы генодерматозов чаще регистрируют ихтиоз (обыкновенный, ламелярный, сцепленный с Х-хромосомой, ихтиозиформенные буллёзные эритродермии), назодигитальный гиперкератоз, семейный дерматомиозит собак, генетические нарушения пигментации (витилиго , лентиго и др.), буллезные эпидермолизы (Эммануэль Бонсиньор).
Ихтиозы – наследственные заболевания, характеризующиеся диффузным нарушением ороговения по типу гиперкератоза и проявляюееся образованием на коже чешуек. Различают ихтиоз обыкновенный (вульгарный), ихтиоз рецессивный Х-сцепленный, ихтиоз ламоллярный, ихтиоз эпидермолитический.
Ихтиоз обыкновенный характеризуется диффузным поражением кожи туловища, конечностей, функциональной недостаточностью эндокринных органов и иммуннодефицитным состоянием.
Тип наследования – рецессивно-доминантный.
Симптомы
. Кожа сухая. Шершавая на ощупь. Выявляют ретенционный гиперкератоз, обусловленный дефектом синтеза кератогеалина, недостатком синтеза филагрина.
Ихтиозам подвержены различные породы собак, особенно терьеры (йоркширский, Джек Рассел, бультерьер, вести).

Степень выраженности кожных изменений различна. Наиболее сильно изменения кожи выражены на разгибательных поверхностях конечностей, подушечек лап, кончика носа. Кожные поражения имеют диффузный или локальный характер. Отмечают скопление десквамированных чешуек, а затем твёрдых корочек. Часто поражаются подушечки лап. Вследствие этого затрудняется движение. Места поражения кожи имеют неприятный прогорклый запах. Животное теряет свой естественный вид, становится уродливым.
Диагноз
ставят по клиническим признакам.
В биопсийном материале кожи при гистологическом исследовании обнаруживают ортокератозный гиперкератоз.
Лечение
. Малоэффективно. Показано применение специальных шампуней, синтетических ретиноидов (тигазон, неотигазон и др.) ориентировочно 0,5-1,0 мг/кг в сутки в течение 2-3 мес. и более.
Назодигитальный гиперкератоз – характеризуется значительным уплотнением носа или подушечек лап. Это заболевание является разновидностью ихтиоза обыкновенного.
При назодигитальном гиперкератозе подушечки лап сильно ороговевают, движение затруднено, появляется хромота. Болеют преимущественно собаки пород ирландского терьера и бордоского дога в раннем возрасте.
Диагноз
ставят по клиническим признакам. Для уточнения диагноза возможно гистологическое исследование биопсийного материала пораженных зон.
Лечение
. Направлено на размягчение уплотнённых зон с помощью водных процедур или применения гелей. Иногда прибегают к хирургическому вмешательству.
Ихтиоз рецессивный Х-сцепленный– заболевание, обусловленное генной мутацией, сопровождаемой отсутствием фермента стероидсульфатазы в эпидермиальных клетках и лейкоцитах. Заболевание характерно только мужскому полу.
Патогенез.
Дефицит фермента стероидсульфатазы приводит к накоплению холестеролсульфата в сыворотке крови и роговом слое, увелтчтвая сцепление клеток и замедляя процесс нормальной десквамации эпидермиса. Холестеролсульфат ингибирует гидроксиметилглутамил-кофермент А-редуктазу – ключевой фермент эпидермального стероидного синтеза.
Симптомы
. у человека заболевание характеризуется поражением кожного покрова в первые месяцы жизни или с самого рождения. Чешуйки на коже большие, тёмные. Гиперкератоз особенно выражен в области разгибательных поверхностей локтевых и коленных суставов. Гистологически выявляют ретенционный гиперкератоз. Для Х-сцепленного ихтиоза характерна так же катаракта, возможны крипторхизм , малые размеры яичек. В сыворотке крови выявляют выраженную холестеринемию. Очевидно по такому же типу развивается Х-сцепленный ихтиоз и у животных.
Ихтиоз ламеллярный– обусловлен недостатком синтеза трансглютаминазы 1. Заболевание проявляется при рождении и развивается в процессе жизни.
«Семейный» дерматомиозит собак– воспаление кожи и мышц чаще встречается у пород колли и шелти молодого возраста. На фоне наследственной предрасположенности провоцирующими этиологическими факторами являются вирусы, травмы, ультрофиолетовое излучение и др.
У больных отмечают депигментацию кожи, кожные сыпи, признаки себореи. Поражение мышц проявляются в напряженной походке, скованности движений, миопатии.
Заболевание дифференцируют от красной волчанки, демодекоза, дерматофитии, буллёзного эпидермолиза. При гистологическом исследовании выявляют вакуолизацию базальных клеток эпидермы, многоочаговый мышечный некроз.
Для лечения показана этиотропная терапия, применение глюкокортикоидов и других средств.
Буллёзные эпидермолизы.
Под буллёзными эпидермолизами понимают наследственные дерматозы, подразделяемые на эпидермолизы буллёзные простые, эпидермолизы

буллёзные сливающиеся, эпидермолизные дистрофии расслоённые базальные. Заболевания встречаются у пуделей, немецких легавых.
Поражения кожи проявляются в виде эрозий, изъязвлений и струпьев, расположенных на внутренней поверхности ушных раковин и других участках кожи. Диагноз подтверждается результатами гистологических исследований биопсийного материала кожи. Выявляют вакуолизацию на уровне базальной мембраны и дермоэпидермальных расслоений.
Лечение
Не эффективное.
К генетическим нарушениям пигментации относятся аномалии: белая шерсть, голубая или гетерохромная радужная оболочка глаз, альбинизм , витилиго, депигментация и др.
Профилактика генодерматозов заключается в ограничении или недопущении случки животных с такой врождённой аномалией.
Альбинизм – врождённое заболевание, характеризующееся отсутствием пигмента кожи, волос, радужной оболочки глаз. Заболевание относится к группе генодерматозов.
Патогенетическая сущность заболевания заключается в блокировании фермента, который необходим для нормального синтеза меланина. В этих условиях меланоциты присутствуют в коже, волосе, оболочках глаз в нормальных количествах, но меланин в них отсутствует, поэтому они не имеют нормального, присущего им цвета.
Волосы, кожа без пигментации, цвет глаз голубой (синдром голубого добермана). Других характерных признаков для этого заболевания не отмечается.
Термин «факоматозы» происходит от греческого слова «фа-кон» – невус.
Факоматозы
– заболевания, характеризующиеся комбинированными невоидными опухолями, гамартомами нескольких органов (кожи, нервной системы и др.). Это моногенные дефекты. Действие гена проявляется еще во внутриутробном развитии, когда на эмбриональной стадии нарушается дифференцировка. Это действие обычно продолжается постнатально. Для всех факоматозов характерно прогредиентное течение.
Наследование преимущественно аутосомнодоминантное.
Кожные симптомы не определяют тяжести заболевания, но имеют большое диагностическое значение.
Туберозный склероз (болезнь Прингля-Бурневиля, эпилойя)
Наследование аутосомно-доминантное, поражаются оба пола, экспрессивность весьма вариабельна, возможна неполная пенетрантность. Популяционная частота- 1:30 000. Классическая триада:
поражение кожи, эпилепсия и умственная отсталость. Течение прогредиентное, но медленное.
Поражения кожи:
1) псевдоаденомы лица (гамартомы с ангиоматозной пролиферацией, наличием сосудистых, соединительнотканных элементов, сально-волосяных структур);
2) ахроматические листовидные пятна;
3) околоногтевые фибромы;
4) соединительнотканные невусы в виде шагреневых бляшек. Псевдоаденомы лица появляются обычно в 4-8-летнем возрасте или несколько позже, не нагнаиваются и не исчезают в отличие от вульгарных угрей, четко отграничены и более плотны.
У взрослых могут образовывать подобие ринофимы. Клинически:
тесно сгруппированные, симметричные многочисленные полушаровидные узелки красноватого цвета с желтоватым или коричневым оттенком. Локализация:
нос, прилегающие щеки, носогубная и подбородочная складки.
Ахроматические листовидные пятна
– самый ранний симптом (может быть с рождения) – хорошо видны в лампе Вуда. Часто имеют неправильные зазубренные очертания. Количество – 4-6, описано до 22 – на спине, пояснице, ягодицах, иногда груди, конечностях. Диаметр – несколько сантиметров.
Околоногтевые фибромы
появляются с возрастом и не у всех. Представляют собой растущие гамартомы, состоящие из коллагеновых волокон и сосудов, обычно множественные.
Соединительнотканные невусы
появляются за счет пролиферации фиброзной ткани – на коже лба, век, поясницы и др. Слегка выступают, желтоватого оттенка с шероховатой поверхностью. Диаметр может быть довольно большим (до 10-15 см).
Нейрофиброматоз (болезнь Реклингаузена)
Популяционная частота 1:3000. Более 2-3 поколений обычно не передается.
Выделяют 2 формы:
периферический и центральный (опухолеподобные симптомы поражения ЦНС) нейрофиброматоз. С рождения имеются «кофейные пятна», а нейрофибромы, изменения костей, диэнцефальные нарушения формируются в дальнейшем.
«Кофейные пятна»
– мелкие лентигоподобные пятна и мягкие нейрофибромы. Типичная локализация – подмышечные складки, промежность. Затем появляются более крупные пятна
(до 2-3 см), может быть диффузная пигментация (например, на спине). Высыпания практически не встречаются на конечностях (обычно – на туловище, шее). У взрослых новые пятна не появляются, в пожилом возрасте некоторые из них могут исчезнуть.
На втором десятилетии и позднее появляются кожные опухоли и опухоли нервных стволов (возможна любая локализация).
Лечение симптоматическое.
Избегать стимулирующих средств, физиотерапии.
Ангиоматозные факоматозы
Синдром Сгердже-Вебера-Краббе:
односторонний сосудистый невус на лице с рождения, поражение сосудов глаз и внутричерепных на стороне поражения.
Синдром Клипиеля
-Треноне: односторонний телеангиэктати- ческий невус, варикозное расширение вен на одной конечности.
Нарушения пигментации
Альбинизм
Заболевание, при котором меланин отсутствует или образуется в недостаточном количестве в коже, волосах, склерах, сетчатке глаза. Количество меланоцитов остается нормальным.
Основная причина – недостаточность тирозиназы.
Тотальный альбинизм.
Возможно Х-сцепленное и аутосомно-рецессивное наследование. Частота возникновения в популяции – 1:10 000. Кожа сухая, розовая, снижено потоотделение, возможен гипотиреоз. Глаза красные, волосы бесцветные. Характерны снижение остроты зрения, рефракции, светобоязнь, блефароконыонктивиты. Из-за повышения чувствительности к УФО развиваются солнечные дерматиты, приводящие к поверхностной атрофии кожи, телеангиэктазиям, позднее к кератозу. Возможно перерождение в рак. Возможно сочетание с глухонемотой, олигофренией, эпилепсией, полидактилией и др.
Субтотальный альбинизм.
Частота возникновения – 1:20 000 (у индейцев – 1:10 000). Наследование аутосомно-рецессивное, редко – аутосомно-доминантное. В основе – нарушение дифференцировки меланобластов.
Частичный альбинизм.
Отсутствует пигмент в отдельных участках кожи. По средней линии лба – аутосомно-доминантное наследование с неполной пенетрантностью, частота возникновения – 1:20 000,1:25 000. На затылке – аутосомно-рецессивное. Другие дефекты отсутствуют. Может быть изолированный альбинизм глаз.
Буллезный эпидермолиз
Буллезный эпидермолиз
представляет собой группу заболеваний, связанных с мутациями. Разные формы этого заболевания связывают с различными генными мутациями. Часть этих форм наследуется аутосомно-доминантно, часть – аутосомно-рецессивно.
Патогенез изучен плохо.
Для всех форм буллезного эпидермолиза характерны нарушения на уровне базальной мембраны.
Клиническая картина весьма вариабельна
. В абсолютном большинстве случаев болезнь проявляется с первых дней жизни: минимальные механические воздействия на кожу приводят к образованию пузырей и эрозий.
Выделяют простые и дистрофические формы буллезного эпидермолиза.
При простых формах базальная мембрана не повреждается и эрозии заживают без образования рубцов. При дистрофических формах отслоение эпидермиса происходит на границе между базальной мембраной и дермой. Повреждение соединительной ткани приводит к формированию рубцов.
Лечение преимущественно симптоматическое.
Принципиально важным является максимально возможное предупреждение травматизации кожи ц слизистых оболочек: щадящий уход за кожей, правильная профессиональная ориентация и т. д.
При образовании эрозий используют наружные эпителизирующие средства. Для улучшения состояния соединительной ткани кожи показаны высокие дозы витамина Е и циклические ретиноиды. При бактериальных осложнениях заболевания используют различные противомикробные средства. При отставании ребенка в физическом развитии назначают общеукрепляющие препараты, анаболики. При развитии различных рубцовых осложнений (контрактур и т. д.) применяется хирургическая коррекция.
Ихтиозы
Ихтиозы
(от греч. ichthy
– рыба) – группа заболеваний, характеризующихся нарушением ороговения. Наиболее часто встречаются вульгарный, Х-сцепленный, ламеллярный к эпидермолитический ихтиозы.
В основе этих заболеваний лежат мутации генов, ответственных за обеспечение процесса ороговения. Для ихтиозов характерен ретенционный гиперкератоз, обусловленный задержкой отшелушивания роговых чешуек. Наследование ихтиозов различно: аутосомно-доминантное, аутосомно-рецессивное или Х-сцепленное.
Наиболее часто встречается вульгарный ихтиоз.
Первые проявления болезни обнаруживаются обычно в возрасте 3-12 месяцев. Мужчины и женщины болеют с одинаковой частотой. Как правило, заболевание наиболее активно проявляется в период полового созревания. В зрелом возрасте обычно сохраняются только сухость кожи и незначительное шелушение.
Выраженность клинических проявлений варьируется. Поражение кожи при вульгарном ихтиозе
диффузное, наиболее выраженное на голенях и предплечьях. Характерны сухость кожи, мелкопластинчатое шелушение, фолликулярный гиперкератоз. Постоянным симптомом является гиперлинейность кожи ладоней и подошв. В половине случаев вульгарный ихтиоз сочетается с атопическим заболеванием (чаще атопическим дерматитом).
Х-сцепленный ихтиоз
чаще поражает мужчин (ген локализуется в одной из половых хромосом). Заболевание проявляется в первые месяцы жизни. На коже ребенка появляются плотно держащиеся крупные темно-коричневые чешуйки. Нередко формируются крупные роговые наслоения. Характерная локализация – кожа волосистой части головы, шеи, туловища, ягодиц, разгибательной поверхности конечностей. Не поражается кожа ладоней, подошв, лица. В отличие от вульгарного ихтиоза не наблюдается улучшение в течении болезни с возрастом.
Ламеллярный ихтиоз (аутосомно-рецессивный тип наследования) проявляется при рождении.
Кожа новорожденного находится в состоянии эритродермии и покрыта желтовато-коричневатой пленкой, напоминающей коллодий. Веки и губы вывернуты (эти симптомы сохраняются всю жизнь). Через несколько дней развивается шелушение, и у некоторых детей (редко) кожа может даже полностью очиститься. В большинстве случаев заболевание сохраняет свои проявления на протяжении всей жизни. В зрелом возрасте обычно усиливается гиперкератоз и становятся менее заметными проявления эритродермии. Ламеллярный ихтиоз нередко сочетается с деменцией.
Эпвдермолитический ихтиоз (буллезная врожденная эритродермия Брока).
Характерно появление дряблых пузырей на фоне яркокрасной кожи новорожденного. Симптом Никольского
положительный. Пузыри быстро вскрываются с образованием эрозий. Нередко состояние утяжеляет присоединение вторичной бактериальной инфекции. В возрасте 3-5 лет выраженность гиперкератоза увеличивается, а количество пузырей уменьшается. Типичная локализация высыпаний -шея, крупные складки, тыл кистей и стоп. Чешуйки темного цвета, линейной формы, плотно прикреплены к коже.
Лечение ихтиозов
зависит от вида заболевания и тяжести его течения. При врожденном ихтиозе не позднее 10 дня жизни назначаются системные глюкокортикостероиды в дозе от 1,5 до 3,5 мг/кг массы тела в сутки (преднизолон). Продолжительность курса – не менее 1 месяца, затем необходимо постепенное снижение дозы препарата.
При всех клинических формах ихтиоза эффективны системные ретиноиды. Однако эти препараты обладают рядом побочных эффектов. Поэтому их назначение при легких формах ихтиоза не оправдано. В таких случаях возможно наружное использование этой группы лекарственных препаратов.
Кроме топических ретиноидов наружно назначаются ожиривающие и смягчающие средства. Показаны солевые, содовые, масляно-молочные и крахмальные ванны. При всех формах ихтиоза рекомендуются санаторное лечение (благотворно влияет влажный и теплый климат), морские купания.
Глава XIX
ГЕНОДЕРМАТОЗЫ
Генодерматозы – наследственные заболевания кожи, насчитывающие несколько сотен нозологических форм, проявляющихся различными патологическими процессами в коже – нарушениями ороговения, дисхромиями и дистрофиями кожи и ее придатков, невоидными и опухолевыми процессами, а также комплексными нарушениями, включающими патологию кожи и нервной системы (факоматозы), эндокринной, костной и других систем организма. Наиболее часто встречаются ихтиоз, кератодермии, буллезный эпидермолиз. Представителем факоматозов является болезнь Реклингхаузена.
ИХТИОЗ
Ихтиоз (син.: кератома диффузная, сауриаз) – наследственное заболевание, характеризующееся диффузным нарушением ороговения по типу гиперкератоза и проявляющееся образованием на коже чешуек, напоминающих чешую рыбы. Различают несколько форм ихтиоза: вульгарный, Х-сцепленный, плода, ихтиозиформная эритродермия.
Ихтиоз вульгарный
– наиболее распространенная форма, составляющая 80-95% от всех форм ихтиоза. Тип наследования аутосомно-доминантный. Заболевание проявляется обычно на 3-м месяце жизни или несколько позже (до 2-3 лет). У больных отмечаются функциональная недостаточность эндокринной системы (щитовидной, половых желез) в комплексе с иммунодефицитным состоянием (снижение активности В– и Т-клеточного иммунитета), склонность к аллергическим заболеваниям (особенно атопическому дерматиту) при низкой сопротивляемости пиококковым и вирусным инфекциям.
Клиническая картина характеризуется диффузным, различной степени выраженности поражением кожи туловища, конечностей в виде наслоений чешуек разных размеров и цвета (от белесоватых до серо-черных), в результате чего кожа становится сухой, шершавой на ощупь. Наиболее сильно изменения кожи выражены на разгибательных поверхностях конечностей, особенно в области локтей и колен, в то время как шея и сгибательные поверхности локтевых и коленных суставов, а также подмышечные ямки не поражены. Характерен также фолликулярный кератоз в виде мелких суховатых узелков с локализацией в устьях волосяных фолликулов диссеминированного характера. Кожа лица в детстве обычно не поражена, у взрослых отмечается шелушение кожи лба и щек. На ладонях и подошвах выражен сетевидный кожный рисунок с изменениями дерматоглифики и небольшим муковидным шелушением. Ногтевые пластинки становятся сухими, ломкими, шероховатыми, деформированными, волосы истончаются и разрежаются. Степень выраженности кожных изменений может быть различной. Абортивный вариант ихтиоза протекает наиболее легко и характеризуется сухостью, небольшой шероховатостью кожи преимущественно разгибательных поверхностей конечностей.
Гистологически выявляют ретенционный гиперкератоз, обусловленный дефектом синтеза кератогиалина. Пролиферативная активность эпидермиса не нарушена. Клинические проявления ихтиоза ослабевают в период полового созревания. Заболевание длится всю жизнь, обостряясь в зимнее время. Нередки конъюнктивит, ретинит, фарингит с субатрофическим поражением носоглотки, отит, риносинусит, хронический мезотимпанит.
Ихтиоз рецессивный Х-сцепленный
выделен из вульгарного ихтиоза на основании генетических исследований. Выявлены случаи делении в коротком плече Х-хромосомы, Х-У-транслокации в кариотипе больных; генная мутация проявляется биохимическим дефектом – отсутствие фермента стероидсульфатазы в эпидермальных клетках и лейкоцитах.
Клиническая картина, развивающаяся в потном объеме только у мальчиков, характеризуется поражением всего кожного покрова, включая кожные складки (30% случаев), в первые месяцы жизни (реже с рождения). Остаются непораженными только ладони и подошвы. У детей в процесс вовлечена кожа волосистой части головы, лица, шеи. С возрастом патологические изменения в указанных зонах ослабевают, а усиливаются изменения кожи в области живота, груди, конечностей. Чешуйки при этой форме ихтиоза большие, темные. Гиперкератоз особенно выражен в области разгибательных поверхностей локтевых и коленных суставов. Клинически данная форма характеризуется коричневато-черным цветом плотно сидящих чешуек, многочисленными мелкими трещинами рогового слоя и крупными (до 1 см) щитками грязно-серого или бурого цвета, отчего кожа напоминает змеиную или панцирь ящерицы. В некоторых случаях клинические проявления напоминают таковые при зульгарном ихтиозе, однако фолликулярный кератоз и изменения кожи ладоней и подошв отсутствуют.
Гистологически выявляют ретенционный гиперкератоз (при нормальном, а не истонченном в отличие от вульгарного ихтиоза зернистом слое). Пролиферационная активность эпидермиса не изменена, но продукция кератогиалина (в отличие от вульгарного ихтиоза) не нарушена. Дефицит фермента стероидсульфатазы приводит к накоплению холестеролсульфата в сыворотке крови и роговом слое, увеличивая сцепление клеток и замедляя процесс нормальной десквамации эпидермиса. Кроме того, холестерол сульфат ингибирует гидроксиметилглутамил – кофермент А-редуктазу – ключевой фермент эпидермального стероидного синтеза.
Для Х-сцепленного ихтиоза характерна также глубокая стромальная катаракта, возможны крипторхизм, малые размеры яичек, полового члена, инфертильность, отставание умственного развития.
В диагностике этой формы ихтиоза, помимо клинической картины и гистологических данных, большое значение имеют результаты биохимического исследования, которые позволяют выявить накопление холестерола в сыворотке крови, коже. Возможна антенатальная диагностика этого вида ихтиоза по определению содержания эстрогенов в моче беременных, количество которых резко снижено вследствие отсутствия фермента арилсульфатазы в плаценте плода, осуществляющей гидролиз предшественников эстрогенов, вырабатываемых надпочечниками плода, что может быть выявлено при амниоцентезе.
Ихтиоз плода (плод Арлекина)
– врожденный ихтиоз, развивающийся в эмбриональном периоде (на IV-V месяце беременности). Тип наследования аутосомно-рецессивный. Генетически гетерогенен, различные фенотипы проявляются отсутствием или присутствием гиперпролиферативных кератинов 6 и 16, профилагрина. Возможно существование мутаций, не совместимых с жизнью, – летальные мутации (в хромосоме 4), что приводит к выкидышу или рождению мертвого плода.
Клиническая картина ихтиоза к моменту рождения ребенка полностью сформирована. Кожа новорожденного сухая, утолщенная, покрыта роговым панцирем, состоящим из роговых щитков серо-черного цвета толщиной до 1 см, гладких или зазубренных, разделенных бороздами и трещинами. В кожных складках поражение напоминает acantosis nigricans. Характерна также диффузная кератодермия ладоней и подошв. Ротовое отверстие нередко растянуто, малоподвижно или, наоборот, резко сужено, напоминает хобот, едва проходимо для зонда. Нос и ушные раковины деформированы, веки выворочены, конечности уродливые (косорукость, контрактуры, синдактилия). Часто наблюдаются тотальная алопеция и дистрофия ногтей, нередко микроофтальмия, микрогирия, катаракта. Большинство детей рождаются мертвыми, остальные умирают вскоре после рождения от изменений внутренних органов, не совместимых с жизнью, истощения, сепсиса.
Гистологически в эпидермисе выявляется диффузный мощный гиперкератоз – роговой слой в 20-30 раз толще всего росткового слоя эпидермиса и содержит много липидов. Зернистый слой утолщен, структура кератогиалиновых гранул не изменена, количество их увеличено, клеточные мембраны утолщены.
Эритродермия ихтиозиформная врожденная
– форма врожденного ихтиоза, выделенная Броком в 1902 г. Различают сухой и буллезный тип. Буллезный тип в дальнейшем чаще стал именоваться эпидермолитическим гиперкератозом (ихтиозом), а эритродермия ихтиозиформная небуллезная врожденная стала многими авторами отождествляться с ламеллярным ихтиозом. Однако биохимические исследования и незначительные признаки клинической картины выявляют некоторые различия.
Ихтиоз ламеллярный
проявляется при рождении ребенка клинической картиной так называемого коллоидального плода. Кожа ребенка при рождении покрасневшая, полностью покрыта тонкой, сухой желтовато-коричневой пленкой, напоминающей коллодий. Такая пленка, просуществовав некоторое время, превращается в крупные чешуйки. С возрастом эритродермия регрессирует, а гиперкератоз усиливается. Поражение захватывает все кожные складки, причем изменения кожи в них часто более выражены. Кожа лица обычно красная, натянутая, шелушится. Волосистая часть головы покрыта обильными чешуйками. Наблюдается повышенная потливость кожи ладоней, подошв, лица.
Волосы и ногти растут быстро (гипердермотрофия), ногтевые пластинки деформируются, утолщаются; отмечаются подногтевой гиперкератоз, диффузный кератоз ладоней и подошв. Характерным проявлением ламеллярного ихтиоза является также эктропион, которому нередко сопутствуют лагофтальм, кератит, фотофобия. Иногда при ламеллярном ихтиозе наблюдается умственная отсталость.
Гистологически: пролиферационный гиперкератоз (иногда с паракератозом), умеренный акантоз, гипертрофия сосочков дермы, средней выраженности хронические воспалительные инфильтраты в верхних слоях дермы. В основе гистогенеза лежит неспособность кератиноцитов образовывать краевую полосу рогового слоя; биохимически выявляется увеличение уровня стерола и жирных кислот в чешуйках кожи.
Сухой тип ихтиозиформной эритродермии
, совпадая практически по клинической картине с ламеллярным ихтиозом, имеет следующие отличия: чешуйки чаще светлые (при ламеллярном ихтиозе более толстые, темные), эритродермия выраженная, вариабельной интенсивности (при ламеллярном ихтиозе средняя), отмечается некоторое разрежение волос на голове (при ламеллярном ихтиозе, кроме этого, возможны аномалии волосяного стержня), эктропион средний (при ламеллярном выраженный; скрученные ушные раковины); гистологически обнаруживается заметное утолщение эпидермиса с паракератозом (при ламеллярном еще и гранулез); биохимически выявляют увеличение содержания п-алканов – ненасыщенных углеводородов, отличающихся гидрофобностью и возможностью влиять на митотическую активность эпидермиса (при ламеллярном ихтиозе – увеличение уровня стерола и жирных кислот в чешуйках кожи).
Ихтиоз эпидермолитический (гиперкератоз эпидермолитический, эритродермия ихтиозиформная буллезная)
– редкая форма врожденного ихтиоза; наследуется по аутосомно-доминантному типу. Заболевание проявляется сразу после рождения ребенка в виде «коллоидального плода». После отторжения пленки кожа новорожденного производит впечатление ошпаренной. Она ярко-красного цвета, с обширными участками отслоения эпидермиса с образованием эрозий и пузырей различной величины, с вялой покрышкой и положительным симптомом отслойки пузыря. Кожа ладоней и подошв утолщена, беловатого цвета, эктропиона нет. В тяжелых случаях процесс сопровождается геморрагическим компонентом (пурпура) и приводит к летальному исходу. В более легких случаях дети выживают. Чаще с возрастом количество пузырей резко уменьшается, а ороговение кожи усиливается неравномерно на разных участках. На 3-4-м году жизни отчетливо выявляется гиперкератоз в виде толстых коричневых веррукозных наслоений. Лицо обычно не поражено, за исключением легкого кератоза носогубных складок; рост волос и ногтей ускорен. На коже туловища может быть гиперкератоз типа игл, почти генерализованный, но неравномерный, сильнее выраженный в области складок кожи, где он принимает вид роговых гребешков. Характерно концентрическое расположение гребешков на разгибательных поверхностях суставов. Периодически на коже появляются пузыри, оставляющие эрозии, количество которых более выражено в первые несколько лет жизни.
Гистологически выявляют эпидермолитический пролиферационный гиперкератоз, акантоз, вакуолизацию цитоплазмы клеток зернистого и шиповатого слоев. Митотическая активность эпидермиса усилена. В основе гистогенеза лежит нарушение образования тонофибрилл, в связи с чем нарушаются межклеточные связи и наблюдается эпидермолиз с образованием щелей и лакун.
Лечение. Ретиноиды (тигазон, неотигазон и др.) из расчета 0,5-1,0 мг/кг в сутки в течение 2-3 мес и более (до 1 года) в зависимости от клинической картины с постепенным снижением дозы. Возможно также использование повторных курсов витаминов А (400 000 ЕД/сут), аевита, С, группы В, биотина. Для нормализации жирового обмена назначают липамид, метионин, рибосан и др. При врожденной ихтиозиформной эритродермии в периоде новорожденности назначают кортикостероидные гормоны (преднизолон из расчета 0,75-3,5 мг/кг в сутки) в комбинации с антибиотикотераиией, анаболическими гормонами, гемодезом, что позволяет значительно ослабить клиническую картину ихтиоза в дальнейшем. Показаны гидропроцедуры: солевые ванны (100 г хлорида натрия или морской соли на ванну) с последующим втиранием в кожу 10% солевою крема на ланолине и рыбьем жире. крахмальные (1 столовая ложка клейстера на ванну), содовые ванны с отрубями, сульфидные, углекислые и др.; талассотерапия, гелиотерапия, иловые и торфяные грязи, УФ-лучи в субэритемных дозах, реПУВА-терапия, иммунотерапия (γ-глобулин и др.). Наружно назначаю)– мази с витамином А (100 000 ЕД на 1 г основы), 0,1% тигазоновый крем, 2% салициловую мазь, 5% с мочевиной, 1-20% мазь с яблочной, лимонной или глюкуроновой кислотой.
КЕРАТОДЕРМИИ
Кератодермия (син. кератоз ладонно-подошвенный) – группа болезней ороговения, характеризующаяся избыточным рогообразованием преимущественно в области ладоней и подошв.
По характеру клинической картины ксратодермии могут быть диффузными, со сплошным поражением всей поверхности ладоней и подошв (кератодермии Унны– Тоста, Меледа, Папийона-Лефе
вра
и др.). и локализованными, при которых участки избыточного ороговения располагаются очагами (кератодермия Бушке-Фишера-Брауэра и др.)
Кератодермия Унны-Тоста (кератома врожденная ладонно-подошвенная)
– распространенная форма наследственной диффузной кератодермии, для которой характерен кератоз ладоней и подошв без перехода на другие участки кожи. Тип наследования аутосомно-доминантный. Заболевание проявляется в первые годы жизни в виде легкого утолщения кожи ладоней и подошв. Постепенно диффузный кератоз нарастает к 4-5 годам, редко позднее К этому возрасту клиническая картина заболевания формируется полностью. Роговые наслоения па ладонях и подошвах (иногда только па подошвах) гладкие, толстые, желтого цвета, с резко очерченным краем, который окружен эритематозным венчиком шириной 1-3 мм. Процесс сопровождается локальным гипергидрозом. Гистологически выявляют ортогиперкератоз, гранулез, акантоз, в дерме – небольшой периваскулярный воспалительный инфильтрат. Волосы, зубы не изменены. Ногти могут быть утолщены (18% случаев), но не дистрофичны. Возможны остеопороз и остеолиз фаланг, деформирующий артроз межфаланговых суставов, осложнение процесса грибковой инфекцией.
Кератодермия Меледа (кератоз наследственный трансградиентный)
– форма наследственной диффузной кератодермии, отличающаяся переходом кератоза с ладонно-подошвенных поверхностей на тыл кистей, стоп, области локтевых, коленных суставов (трансградиентный кератоз). Описана впервые среди кровных родственников населения острова Меледа. Тип наследования обычно аутосомно-рецессивный. Первые проявления болезни возникают в детском возрасте в виде стойкой эритемы с шелушением кожи ладоней и подошв. В дальнейшем ороговение кожи усиливается, и к 15-20 годам на ладонях и подошвах видны массивные роговые наслоения желто-коричневого цвета, лежащие компактными пластинами, эритема сохраняется лишь в виде фиолетово-лилового ободка шириной несколько миллиметров по периферии очага. Гистологически выявляют гиперкератоз, иногда акантоз, в дерме – небольшой воспалительный лимфогистиоцитарный инфильтрат. Характерен локальный гипергидроз, поверхность очагов кератоза обычно влажная, с черными точками выводных протоков потовых желез. Роговые наслоения переходят на тыльную поверхность кистей, стоп, области локтевых и коленных суставов, на их поверхности образуются болезненные глубокие трещины (особенно в области пяток). Характерны сочетание с атопическим дерматитом, осложнения процесса пиококковой инфекцией, дистрофия ногтей с их резким утолщением или койлонихией. Могут отмечаться изменения на электроэнцефалограмме, умственная отсталость, синдактилия, складчатый язык, готическое небо.
Кератодермия Папийона-Лефевра (Папийона-Лефевра синдром)
– наследственная диффузная кератодермия, сочетающаяся с пародонтозом и пиогенными инфекциями кожи и десен. Тип наследования аутосомно-рецессивный. У больных отмечаются снижение функции щитовидной и поджелудочной железы, нарушение функциональной активности лейкоцитов, снижение фагоцитарной активности нейтрофилов и чувствительности Т– и В-лимфоцитов к митогенам. Клиническая картина проявляется обычно в возрасте от 1 года до 5 лет (чаще на 2-3-м году жизни) в виде эритемы ладоней и подошв, покрывающихся роговыми наслоениями, интенсивность которых постепенно усиливается. Участки кератоза нередко выходят за пределы ладонно-подошвенных поверхностей на тыл кистей и стоп, область пяточного (ахиллова) сухожилия, коленных и локтевых суставов. Характерен локализованный гипергидроз. Гистологически выявляют гиперкератоз, нерегулярный паракератоз, в дерме – небольшой воспалительный инфильтрат. В клетках рогового и зернистого слоев обнаруживают липидоподобные вакуоли, нарушение структуры тонофибрилл и кератогиалиновых гранул. Ногти нередко дистрофичны (тусклые, ломкие), волосы не изменены. В возрасте 4-5 лет в результате персистирующего гингивита развивается прогрессирующий пародонтоз с образованием гнойных альвеолярных карманов, воспалением и дистрофией альвеолярных отростков с преждевременным кариесом и выпадением зубов, аномалией их развития. Возможны кальцификация твердой мозговой оболочки, арахнодактилия, акроостеолиз.
Кератодермия диссемицированная Бушке-Фишера-Брауэра (кератоз точечный рассеянный Бушке-Фишера)
– наиболее распространенная форма очаговой наследственной кератодермии. Тип наследования аутосомно-доминантный. Первые симптомы болезни появляются в пубертатном периоде или несколько позже (от 15 до 30 лет). На коже ладоней, подошв и сгибательной поверхности пальцев появляются роговые узелки – «жемчужины» величиной от 2 до 10 мм в диаметре, которые превращаются в плотные роговые желтовато-коричневые пробки с кратерообразным краем. При отторжении центральных роговых масс остается кратерообразное углубление. Потоотделение не нарушено. Гистологически выявляют гиперкератоз с паракератозом в центральной части, небольшой акантоз, в дерме – незначительный периваскулярный воспалительный инфильтрат.
Диагноз кератодермии основывается в основном на клинических данных. Дифференциальный диагноз проводят с различными формами кератодермии, псориазом, дисгидротической экземой.
Лечение: ретиноиды (тигазон, неотигазон и др.) – по 0,5 мг/кг в сутки в течение нескольких недель, аевит, ангиопротекторы (теоникол, трентал и др.); наружно: кератолитические мази (20% салициловая марь. мазь Ариевича). солевые ванны, фонофорез с витамином А.20% димексид, лазеротерапия. При кератодермии Папийона-Лефевра лечение начинают с антибиотикотерапии и санации полости рта.
БУЛЛЕЗНЫЙ ЭПИДЕРМОЛИЗ
Эпидермолиз буллезный (син. пузырчатка наследственная Брока) – группа наследственных буллезных дерматозов, характеризующихся образованием пузырей на коже и слизистых оболочках, возникающих при незначительной травматизации или спонтанно. Выделяют несколько основных типов буллезного эпидермолиза на основе особенностей механизма образования пузыря и клинической картины: простой, соединительный и дистрофический
. В пределах этих типов описано более 10 различных форм заболевания.
Эпидермолиз буллезный простой
характеризуется образованием внутриэпидермальных пузырей в результате дезинтеграции и цитолиза кератиноцитов без признаков рубцевания, атрофии и образования милиумов. Тип наследования аутосомно-доминантный.
Клиническая картина. Первые признаки заболевания обычно проявляются на 1-м году жизни, иногда могут быть уже при рождении ребенка. На месте легкой травматизации, чаще в области кистей, стоп, спины, локтевых и коленных суставов, затылочной области, на неизмененной коже появляются пузыри различных размеров (от 0,5 до 7 см и более) с плотной покрышкой и прозрачным содержимым. Симптом Никольского отрицательный, акантолитические клетки в содержимом пузыря отсутствуют. Через несколько днем пузыри вскрываются, образуя эрозии, покрывающиеся корками и быстро эпителизирующиеся, не оставляя рубцовых изменении кожи или атрофии. Пузырей обычно больше в теплый период года при выраженном гипергидрозе. С возрастом поражения локализуются в основном на конечностях, особенно на стопах и кистях, чему способствует большая травматизация этих участков кожи. тесная, плохо подобранная обувь, а также на участках тесного прилегания одежды. Пузыри появляются на протяжении всей жизни, но в постпубертатный период их количество уменьшается. Слизистые оболочки, ногти не поражаются или их изменения минимальны. Общее состояние больного не изменяется.
Возможна пренатальная диагностика этой формы заболевания по высокому содержанию в сыворотке крови беременной α-фетопротеина во II триместре. Эпидермолиз буллезный простой летний Вебера-Кокейна
– абортивная локализованная форма эпидермолиза буллезного простого. Характеризуется образованием пузырей на коже кистей и стоп лишь в летнее время года при выраженном ладонно-подошвенном гипергидрозе.
Эпидермолиз буллезный соединительный
характеризуется образованием подэпидермальных пузырей за счет поражения lamina lucida эпидермо-дермального соединения, расположенной между плазматической мембраной базальных кератиноцитов и базальной мембраной кожи, и развитием атрофических изменений кожи в очагах поражения. Возможна пренатальная диагностика с помощью биопсии кожи 18-недельного плода на основании выявления указанных изменений. Тип наследования аутосомно-рецессивный.
Клиническая картина. Процесс характеризуется появлением пузырей и эрозий уже при рождении ребенка или вскоре после него. В течение нескольких дней процесс генерализуется. Основная локализация высыпаний – кожа груди, головы, слизистые оболочки рта, гортани, трахеи. Хотя кожа кистей и стоп не изменена, ногтевые пластинки дистрофичны, развиваются анонихия, акроостеолиз. Образующиеся на месте пузырей эрозивные поверхности заливают медленно, оставляя участки атрофии кожи. Рубцов и милиумов нет. Многие дети умирают в первые месяцы жизни от сепсиса, анемии.
Эпидермолиз буллезный дистрофический
характеризуется образованием пузырей вследствие дерматолиза – гибели коллагеновых фибрилл в сосочковом слое дермы ниже lamina densa. Формируются эрозивно-язвенные поверхности, заживающие рубцами, характерны также образование милиумов, изменение ногтей, волос, зубов и другие аномалии.
Эпидермолиз буллезный дистрофический рецессивный генерализованный (эпидермолиз буллезный дистрофический полидиспластический)
отличается образованием пузырей в сосочковом слое дермы и результате дерматолиза – лизиса коллагеновых фибрилл с фагоцитозом их макрофагам и разрушениями ниже lamina densa. Патологический процесс связывают с увеличением уровня и активности фермента коллагеназы, разрушающей основной компонент опорных коллагеновых фибрилл – коллаген VII (коллагенолиз). Возможна пренатальная диагностика болезни по результатам биопсии кожи плода на 21– й неделе развития и выявления описанных ранее изменений.
Первые признаки заболевания появляются уже при рождении (60% больных) или в первые недели жизни. Крупные пузыри, нередко с гемморрагическим содержимым, возникают спонтанно на любом участке кожного покрова и слизистых оболочек. Обширные длительно не заживающие эрозивно-язвенные поверхности, образующиеся при их вскрытии, затрудняют уход и вскармливание новорожденных. Симптом эпидермальной отслойки положительный. На эрозивно-язвенных, нередко кровоточащих, болезненных участках развиваются вегетации. Заживление их происходит медленно, с формированием уродствующих атрофических рубцов. Рубцовые изменения пищевода, глотки, слизистой оболочки рта могут затруднять прием пищи, облитерировать выводные протоки слюнных желез, ограничивать подвижность языка и привести к развитию лейкоплакии. Поражения глаз в виде эрозивно-язвенного кератита с последующим рубцеванием приводят к потере зрения, рубцовому эктропиону, облитерации протоков слезных желез. Наблюдаются также акроцианоз, склеродермоподобные изменения кожи кистей, стоп с формированием сгибательных контрактур суставов, акроостеолизом и характерной деформацией кистей по типу «варежки» в результате срастания и деформации пальцев. Характерна также дистрофия ногтей, волос, зубов. Возможны нарушения эндокринной (гипофункция щитовидной железы, гипофиза), нервной (эпилепсия, отставание умственного развития) систем. Отмечается высокая летальность в раннем детском возрасте от сепсиса, анемии, нарушения питания, в более старшем возрасте – от злокачественных новообразований кожи, пищевода, органов полости рта.
Эпидермолиз буллезный дистрофический доминантный (эпидермолиз буллезный дистрофический гиперпластический)
характеризуется образованием пузырей в дерме (дерматолиз) ниже lamina densa за счет гибели опорных коллагеновых фибрилл; возможна пренатальная диагностика (по аналогии с дистрофическим полидиспластическим буллезным эпидермолизом). Тип наследования аутосомно-доминантный. Первые проявления болезни появляются в раннем детском возрасте или несколько позднее (4-10 лет). Пузыри возникают после незначительной травмы, чаще в области конечностей. Они напряженные, плотные, с серозным или геморрагическим содержимым: вскрываясь, образуют эрозивно-язвенные поверхности, заживающие медленно с образованием мягкой или келоидоподобной рубцовой атрофии вначале розового, затем белого цвета. В области суставов на месте пузырей (формируются обширные поля поражения в виде рубцовой ткани с множеством эпидермальных кист (милиумы). Симптом эпидермальной отслойки положительный. Ногти, вовлеченные в процесс, утолщены, дистрофичны. Слизистые оболочки поражаются редко. Волосы, зубы и общее развитие обычно не изменяются, однако часто отмечается ассоциация с ихтиозом, фолликулярным кератозом, гипертрихозом. Диагноз буллезного эпидермолиза основывается на клинических и гистологических данных. Возможна пренатальная диагностика заболевания. Дифференциальный диагноз в раннем детском возрасте проводят с эпидермолитическим ихтиозом, при котором доминирует кератоз; эпидемической пузырчаткой новорожденных, для которой характерно острое начало с лихорадкой, интоксикацией и воспалительными пузырями в результате некротических процессов в эпидермисе, вызванных стафилококком. У детей более старшего возраста некоторые формы буллезного эпидермолиза дифференцируют от доброкачественного буллезного пемфигоида. который отличается линеарным отложением IgА вдоль назальной мембраны. Антитела к lamina densa, VII типу коллагена, пемфигоидные и др. помогают установить характер дефекта и уточнить диагностику.
Лечение симптоматическое. При простои форме буллезного эпидермолиза важна защита кожи от травматизации, воздействия высокой температуры, тесных одежды и обуви, а также от присоединения вторичной инфекции. Внутрь назначают витамины (А, группы В, С), фитин; пузыри вскрывают и пропитывают (не срывая покрышки) анилиновыми красителями, мазями с антибиотиками, эпителизирующими мазями. При соединительном и дистрофическом эпидермолизе, помимо указанных средств, в тяжелых случаях назначают кортикостероидные препараты (1-3 мг/кг в сутки) в комбинации с антибиотиками, препараты железа (тардиферон и др.), переливания эритроцитарной массы (при анемии), сердечные средства, для ингибирования коллагеназы используют фенитоин.
БОЛЕЗНЬ РЕКЛИНГХАУЗЕНА
Болезнь Реклингхаузена (нейрофиброматоз I типа) – наследственный нейрокутанный факоматоз. Характеризуется развитием множественных нейрофибром, неврином и другими дефектами развития экто– и мезодермы.
Этиология и патогенез. Обусловлен мутантным аутосомно-доминантным геном, локализованным в хромосоме 17, имеющим 100% пенетрантность, благодаря чему заболевание проявляется у всех детей начиная с периода новорожденности до 5-летнего возраста.
Клиническая картина.
Изменения кожи характеризуются множественными нейрофибромами, пятнами цвета кофе с молоком и типа веснушек. Вначале обычно появляются округлые, резко контурированные (цвета кофе с молоком) пятна диаметром от 0,5 до 10 см и более, обусловленные скоплением меланоцитов. Позднее, чаще в подмышечных ямках, наблюдаются мелкие пигментные пятна. Нейрофибромы – доброкачественные опухоли, развиваются из леммоцитов периферических нервов и являются наиболее типичным признаком заболевания. Они появляются обычно позднее (через несколько месяцев или лет, иногда к периоду полового созревания и представляют собой мягкие лилово-розовые, куполообразно возвышающиеся или сидящие на ножке округлые диаметром от нескольких миллиметров до нескольких сантиметров образования. От легкого надавливания многие опухоли втягиваются в кожу (симптом «кнопки от звонка»). Наибольшее количество (возможно, сотни) расположено на туловище (особенно на груди, спине, пояснице). Вдоль периферических нервов могут пальпироваться мягкие фиброзные узелки. Возможно развитие плексиформной невромы – диффузной опухоли, идущей вдоль тройничного нерва или других нервов и состоящей из плотных извилистых тяжей. У 5-10% больных папиломатозные опухоли образуются на внутренней поверхности щек, языке, губах. Абортивная форма болезни Реклингхаузена, характеризующаяся лишь пигментными пятнами, называется синдромом Лешке. Нередко развиваются кифосколиоз, кардиореспираторная патология; нередко снижен интеллект, отставание физического развития, эндокринные нарушения (гинекомастия; гиперпаратиреоидизм и др.), у 40% больных выявляют опухоли ЦНС.
Лечение. Хирургическое иссечение отдельных опухолей.
Наиболее сильно изменения кожи выражены на разгибательных поверхностях конечностей, особенно в области локтей и колен, в то время как шея и сгибательные поверхности локтевых и коленных суставов, а также подмышечные ямки не поражены. Характерен также фолликулярный кератоз в виде мелких суховатых узелков с локализацией в устьях волосяных фолликулов диссеминированного характера. Кожа лица в детстве обычно не поражена, у взрослых отмечается шелушение кожи лба и щек. На ладонях и подошвах выражен сетевидный кожный рисунок с изменениями дерматоглифики и небольшим муковидным шелушением.
В зависимости от вида и степени образования чешуек различают несколько клинических вариантов вульгарного ихтиоза: ксеродермия-абортивный вариант ихтиоза, наиболее легко протекающий, характеризующийся сухостью, небольшой шероховатостью кожи преимущественно разгибательных поверхностей, конечностей. Кожа легко раздражается, особенно при мытье водой с мылом, и предрасположена к экзематизации; ихтиоз простой, при котором поражение охватывает всю кожу.
- Чешуйки мелкие, центральная часть их плотно прикрепляется к основанию;
- ихтиоз блестящий отличается прозрачностью и нежностью чешуек, располагающихся в виде мозаики, главным образом на конечностях;
- ихтиоз белый-чешуйки белые, асбестовидные, создающие впечатление обсыпанной мукой кожи;
- ихтиоз змеевидный-чешуйки крупные серовато-коричневые,напоминающие змеиный покров.
В настоящее время все они расцениваются как вульгарный ихтиоз различной степени выраженности. Гистологически выявляют ретенционный гиперкератоз, обусловленный дефектом синтеза кератогиалина.Пролиферативная активность эпидермиса не нарушена. Нарушен процесс отторжения клеток, что может быть связано с цементирующим действием гликозаминогликанов.
Ногтевые пластинки становятся сухими, ломкими, шероховатыми, деформированными, волосы могут истончаться и разрежаться. Клинические проявления ихтиоза ослабевают в период полового созревания. Заболевание длится всю жизнь, обостряясь в зимнее время. Отмечаются функциональная недостаточность эндокринной системы(щитовидной, половых желез) в комплексе с иммунодефицитным состоянием, выраженная склонность к аллергическим заболеваниям при низкой сопротивляемости пиококковым и вирусным инфекциям. Нередки конъюктивит, ретинит, фарингит с субатрофическим поражением носоглотки, отит , риносинусит, хронический мезотимпанит.
Лечение у косметолога-дерматолога в Москве
Косметолог-дерматолог в нашей клинике “ЕВРОМЕД С” в Москве использует новейшие методики терапии дерматологических патологий.
Лекарственное лечение
— препараты нового поколения, целенаправленно действующие на источник проблемы. Правильно способствуют быстрому разрешению патологического кожного процесса, избавляют от боли, зуда, восстанавливают обменные процессы, укрепляют иммунную систему.
Аппаратная терапия
— эффективные аппаратные технологии помогают быстро и надолго избавить от эстетических проблем. Мезотерапия, лазерное лечение, криотерапия — это лишь несколько методик, которые с успехом используются в клинике Москвы.
Радиоволновая хирургия
— для удаления новообразований мы применяем самую безопасную технологию бесконтактного иссечения тканей. Она гарантирует скорейшее заживление покровов, отсутствие шрамов и рубцов, сокращает риск появления папиллом на том же месте.
Залог успеха любого лечения — индивидуальный подход к каждому пациенту. Врач дерматолог в Марьино
составляет программу терапии в соответствии с особенностями кожи пациента, историей его прошлых заболеваний. Таким образом, достигается максимальная эффективность лечения, долговременный эффект эстетических процедур.
Более подробную информацию о лечении и его стоимости Вы можете получить по телефонам или во время личной консультации у косметолога-дерматолога.
|
|
|
| № | Название услуги | Стоимость |
|---|---|---|
| 1. | Консультация дерматолога с осмотром |
1000 руб. |
| 2. | Вторичный прием дерматолога | 800 руб. |
| 3. | Удаление моллюск Контагиозный моллюск | 500 руб. |
| 4. | Удаление родинок, папиллом, на теле (радиоволновый метод) от 0,5 до 1 см | 500 руб. |
| 5. | Внутривенное УФО крови (“Матрикс-ВЛОК”) | 1200 руб. |
| 6. | Удаление родинок, удаление папиллом, в области лица, шеи, декольте (радиоволновый метод) от 0,5 до 1 см | 1500 руб. |
| 7. | Удаление родинок, папиллом, бородавок в области лица, шеи, декольте (радиоволновый метод) свыше 1 см | 1300 руб. |
| 8. | Удаление кандилом 1 ед (радиоволновый метод) | 1000 руб. |
| 9. | Удаление бородавок | 1000руб. |
| 10. | Курс лечения псориаза: кожных покровов, волосистой части головы, ладонно-подошвенной формы (без стоимости медикаментов) | 2000- руб. |
| 11. | Курс лечения онихомикозов (грибковых поражений) (без стоимости медикаментов) | 3000руб. |
| 12. | Курс лечения микроспорий волосистой части головы и гладкой кожи (без стоимости медикаментов) | 2000руб. |
| 13. | Курс лечения разноцветного лишая (без стоимости медикаментов) | 2000 руб. |
| 14. | Удаление образований на подошве, на ладони | 1000-1500 руб. |
| 15. |